Главная | О сайте | Задачи | Проекты | Результаты | Диверсификация | Новости | Вопросы | История | Информация | Ссылки
Пятница, 01.11.2024
Космическая погода на текущий час
Космическая погода на текущий час
Вход в систему не произведен
Войти / Регистрация
Войти / Регистрация
Секция Совета РАН по космосу
< Лишний вес молодой звезды: БОРЬБА С ПАРАДОКСОМ
Сравнение геномов 29 млекопитающих проливает свет на эволюцию человека
Международная команда генетиков сообщила о завершении шестилетнего проекта по прочтению полных геномов 29 видов плацентарных млекопитающих. Сравнительный анализ прочтенных геномов позволил картировать 3,6 млн. функциональных (консервативных) участков ДНК, находящихся под действием очищающего отбора.
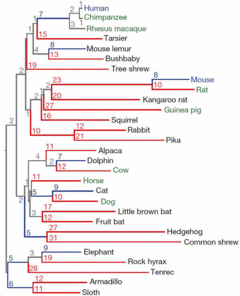
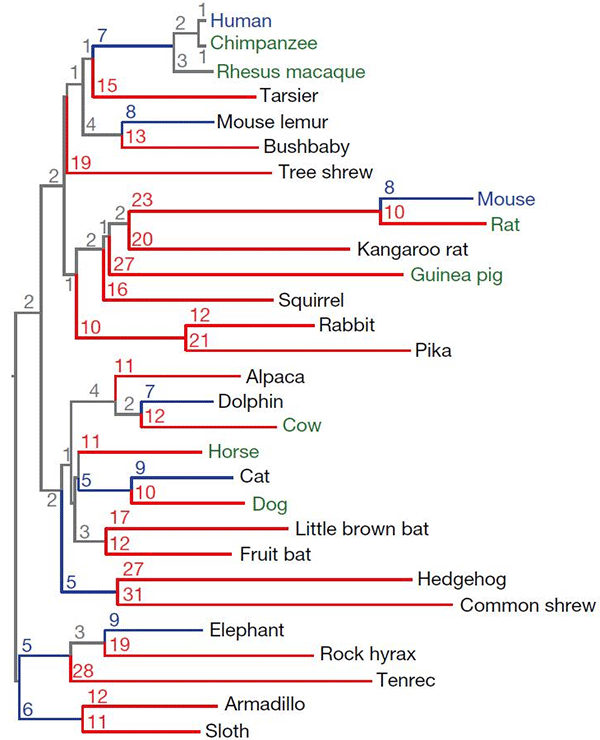
- Эволюционное дерево 29 видов плацентарных млекопитающих, чьи геномы анализируются в рассматриваемой работе. Синим выделены полностью прочтенные геномы, зеленым — высококачественные, черным — менее качественные «черновики». Цифрами показано количество нуклеотидных замен (на сотню) для каждой ветви дерева. Красным выделены ветви с более чем десятью заменами на сотню нуклеотидов. Рисунок из обсуждаемой статьи в Nature
Эти участки, мутации в которых не являются нейтральными, составляют примерно 5,5% генома у плацентарных. Около трети из них соответствуют белок-кодирующим последовательностям, остальные 2/3 — регуляторным последовательностям, расположенным в интронах и межгенных промежутках. Выявлено 280 000 регуляторных участков, происходящих из фрагментов «одомашненных» мобильных генетических элементов, а также 563 участка, эволюция которых шла ускоренными темпами у предков человека после их отделения от предков шимпанзе. Работа представляет собой важный шаг на пути к пониманию смысла прочтенных генетических текстов.
Прошло уже около десятилетия с момента первого, «чернового» прочтения человеческого генома (см. Проект «геном человека»), а работе по его анализу и интерпретации еще не видно конца. Первоочередная задача, стоящая перед исследователями, состоит в выявлении всех «осмысленных», функциональных участков генома и в выяснении их функций.
Главный метод, применяемый для массового выявления значимых участков генома, основан на анализе внутри- и межвидовой генетической изменчивости. Пониженный уровень внутри- и межвидовых различий в каком-то фрагменте генома свидетельствует о том, что большинство мутаций, появлявшихся в этом фрагменте, оказывались вредными и отбраковывались отбором. Стало быть, этот фрагмент зачем-то нужен. «Мусорная ДНК», не выполняющая никаких полезных функций, накапливает мутации свободно, и поэтому соответствующие участки геномов отличаются повышенной вариабельностью.
Ранее человеческий геном уже анализировали таким способом, используя для сравнения геномы трех других плацентарных млекопитающих: мыши, крысы и собаки. Было показано, что не менее 5% генома человека находится «под отбором», причем 1,5% — это белок-кодирующие участки, а остальные 3,5% — некодирующие, предположительно регуляторные. Однако анализ, основанный на сравнении четырех геномов, хоть и позволил приблизительно оценить долю функциональных участков в геноме, но не позволил с достаточной точностью локализовать большую часть этих участков.
Дело в том, что достоверность выводов при таком анализе напрямую зависит от суммарной длины ветвей эволюционного дерева сравниваемых видов, измеряемой средним количеством нуклеотидных замен на сайт (то есть на нуклеотид). Чем больше эта величина, тем ниже вероятность того, что тот или иной фрагмент ДНК остался одинаковым у сравниваемых видов в силу простой случайности (а не потому, что на него действовал очищающий отбор). Для четверки «HMRD» (human, mouse, rat, dog — человек, мышь, крыса, собака) суммарная длина ветвей эволюционного дерева составляет 0,68 замен на сайт. При этом вероятность того, что отдельный нуклеотид, не находящийся под действием отбора, окажется у этой четверки одинаковым, близка к 0,5, а для отрезка длиной в 12 нуклеотидов эта вероятность составляет 10–3.
Чтобы повысить надежность результатов, большая международная команда генетиков в 2005 году приступила к выполнению проекта по прочтению геномов множества видов плацентарных млекопитающих, представляющих все основные ветви эволюционного дерева этой группы животных. За шесть лет работы ученым удалось получить «черновые» последовательности для 29 видов (геномы девяти из них опубликованы ранее, 20 публикуются впервые).
Суммарная длина ветвей дерева, построенного для этих 29 видов (см. рисунок) — около 4,5 замен на сайт. Соответственно, резко уменьшилась вероятность получения ложно-положительных результатов при выявлении участков, контролируемых очищающим отбором. Для отдельного нуклеотида эта вероятность теперь составляет менее 0,02, для отрезка длиной 12 нуклеотидов — 10–25.
Большинство геномов прочтены пока только «вчерне», то есть не очень качественно. Количество ошибочно прочтенных нуклеотидов в них, по оценке авторов, составляет 1–3 на тысячу. Это, однако, примерно в 50 раз меньше среднего уровня нуклеотидных различий между видами. Поэтому полученные данные вполне пригодны для содержательного анализа.
Авторы пришли к выводу, что примерно 5,36–5,44% человеческого генома находится «под отбором». Это мало отличается от прежних оценок, основанных на четверке HMRD, однако теперь удалось большинство этих консервативных (функциональных) участков еще и локализовать, то есть установить их положение на хромосомах. Всего найдено 3,6 миллиона консервативных участков ДНК. Средняя длина участка — 36 нуклеотидов, тогда как консервативные участки, выявленные ранее на четверке HMRD, имеют среднюю длину 123 нуклеотида. Новые данные позволили локализовать множество маленьких функциональных участков (таких, например, как сайты связывания транскрипционных факторов), которые невозможно было выловить, имея только четыре генома для сравнения.
Авторы сопоставили полученные данные с имеющейся информацией по внутривидовой генетической вариабельности Homo sapiens. Оказалось, что эти массивы данных прекрасно согласуются друг с другом. Те участки генома, которые мало отличаются у разных видов плацентарных (то есть медленно меняются в ходе эволюции), в пределах человеческой популяции тоже имеют пониженную вариабельность, и наоборот: те участки, которые у разных людей могут сильно отличаться друг от друга, у других плацентарных тоже изменчивы. Более того, многие сайты с ограниченной «эволюционной пластичностью» (например, позиции, в которых может стоять нуклеотид Г или Т, но не А и не Ц), варьируют одинаковым образом как внутри человеческой популяции, так и у разных видов плацентарных. Это значит, что очищающий отбор, действовавший на геномы различных плацентарных, продолжал схожим образом действовать и на геномы ближайших предков современного человечества.
Примерно 30% из найденных 3,6 миллионов консервативных участков расположены в белок-кодирующих последовательностях или в их ближайших окрестностях. Еще 30% приурочены к интронам, остальные 40% — к межгенным промежуткам. Авторы обнаружили 3788 новых экзонов, а также свыше 10 000 участков кодирующих последовательностей с резко пониженной частотой синонимичных замен. В этих участках оказываются вредными и отбраковываются отбором не только значимые замены, меняющие аминокислоту в белке, но и синонимичные замены, не влияющие на структуру кодируемого белка и затрагивающие только структуру матричной РНК. Как правило, это характерно для участков, важных для регуляции сплайсинга или связанных с регуляцией активности генов при помощи микроРНК. Особенно много таких участков обнаружено в HOX-генах (см. Новое в науке о знаменитых Hox-генах, регуляторах развития, «Элементы», 10.10.2006). Найдено много новых РНК-переключателей (см. Сложные РНК-переключатели — новый механизм регуляции генов, «Элементы», 18.10.2006), генов функциональных РНК, сайтов связывания транскрипционных факторов и других регуляторных элементов. В общей сложности удалось найти ту или иную предположительную функцию примерно для 60% обнаруженных консервативных элементов. Зачем нужны остальные 40%, пока неизвестно.
Проведенное авторами картирование консервативных участков генома человека должно помочь медицинским генетикам в поисках мутаций (полиморфизмов), связанных с различными наследственными заболеваниями. К настоящему времени выявлено большое количество однонуклеотидных полиморфизмов, наличие которых коррелирует с теми или иными заболеваниями. Однако такая корреляция далеко не всегда обозначает причинно-следственную связь. С заболеванием может коррелировать несколько полиморфизмов, расположенных по соседству (например, в окрестностях одного и того же гена). При этом подлинной причиной болезни может быть только один из них, а остальные коррелируют с ней просто потому, что находятся рядом (см. неравновесие по сцеплению). Именно для расшифровки таких случаев и нужна подробная карта консервативных участков. Полиморфизм, находящийся в пределах консервативного (то есть функционально важного) участка, с гораздо большей вероятностью является причиной заболевания, чем полиморфизм, расположенный по соседству в участке, не находящимся под действием очищающего отбора. Авторы приводят конкретный пример расшифровки одного такого случая. Два полиморфизма, расположенные неподалеку друг от друга в межгенном участке, разделяющем хокс-гены HOXB1 и HOXB2, коррелируют с наследственными нарушениями развития зубов. При этом один из полиморфизмов находится в консервативной области, а другой — нет. Авторы показали, что первый из этих полиморфизмов нарушает структуру широко распространенного у млекопитающих регуляторного «мотива» — последовательности ДНК, к которой, по всей видимости, должен прикрепляться какой-то белок — регулятор транскрипции. Скорее всего, именно этот полиморфизм является подлинной причиной аномальных зубных фенотипов.
Среди обнаруженных 3,6 млн. консервативных участков свыше 280 000 происходят из фрагментов «одомашненных» мобильных генетических элементов (МГЭ). До сих пор у плацентарных было выявлено лишь около 10 000 таких случаев. Таким образом, исследование расширило представления о характере и масштабах «молекулярного одомашнивания» — процесса, в ходе которого фрагменты эгоистических, самопроизвольно перемещающихся по геному участков ДНК оказываются полезными для организма и превращаются в функциональные элементы генома (см. Прочтение генома опоссума доказало ключевую роль транспозонов в эволюции млекопитающих, «Элементы», 13.05.2007).
Авторы оценили также масштабы действия положительного отбора полезных мутаций. Основным критерием служило соотношение значимых и незначимых нуклеотидных замен в белок-кодирующих последовательностях. Анализ проводился на уровне отдельных кодонов (троек нуклеотидов, каждая из которых кодирует одну аминокислоту). Получилось, что 84,2% кодонов в ходе эволюции плацентарных находились под действием сильного очищающего отбора (мутации, меняющие кодируемую аминокислоту, в большинстве случаев отбраковывались отбором), а 2,4% кодонов часто оказывались под действием положительного отбора, то есть значимые мутации этих кодонов оказывались полезными и закреплялись.
Выявлено также 563 консервативных участка генома, которые подвергались ускоренной эволюции под действием положительного отбора у предков человека после их отделения от предков шимпанзе. Такие участки называются «human-accelerated regions» (HARs). Ранее было известно только 202 таких участка. Еще 577 участков подвергались ускоренной эволюции у приматов в целом (primate-accelerated regions, PARs). Среди генов, расположенных по соседству с HARs и PARs, преобладают гены, участвующие в обмене сигналами между клетками, иммунной защите, регуляции эмбрионального развития и роста аксонов.
Исследование представляет собой важный шаг на пути к пониманию смысла прочтенных генетических текстов. Можно надеяться, что скоро это понимание станет еще более полным, потому что в ближайших планах специалистов по сравнительной геномике — прочтение полных геномов 10 000 видов позвоночных (см.: Genome 10K Project).
Источник: Kerstin Lindblad-Toh, Manuel Garber, Or Zuk, Michael F. Lin, Brian J. Parker, Stefan Washietl, Pouya Kheradpour, Jason Ernst, Gregory Jordan, Evan Mauceli, Lucas D. Ward, Craig B. Lowe, Alisha K. Holloway, Michele Clamp, Sante Gnerre, Jessica Alföldi, Kathryn Beal, Jean Chang, Hiram Clawson, James Cuff, Federica Di Palma, Stephen Fitzgerald, Paul Flicek, Mitchell Guttman, Melissa J. Hubisz, et al. A high-resolution map of human evolutionary constraint using 29 mammals // Nature. 2011. V. 478. P. 476–482.
- Ссылки:
Комментарии
Комментарии
Магнетары
Магнетарами называются нейтронные звезды, (см. Звезды, нейтронные) обладающие мощнейшим магнитным полем... [далее]
Сайт разработан и поддерживается лабораторией 801 Института космических исследований Российской академии наук.
Подбор материалов - Н.Санько
Полное или частичное использование размещённых на сайте материалов
возможно только с обязательной ссылкой на сайт Секция Солнечная система Совета РАН по космосу.